پارس اتیلن کیش به کیفیت متعهد است
با پارس اتیلن کیش کیفیت را تجربه کنید
پارس اتیلن کیش تولید
کننده با کیفیت ترین لوله ،اتصالات ومنهول پلی اتیلن در ایران
شرکت پارس اتیلن کیش هیچ نماینده ای در سطح ایران ندارد و فروش لوله و اتصالات پلی
اتیلن از طریق دفتر مرکزی با ارائه گواهینامه معتبر انجام میپذیرد.
لوله های کاروگیت پارس اتیلن کیش، تحت لیسانس DROSSBACH آلمان تولید میگردد
از شرکت هایی خرید کنید که با کارکنان ,مشتریان و محیط زیست با احترام
رفتار میکنند
پارس اتیلن کیش نامی که می
شناسید و به آن اعتماد دارید
اروپائی ها هم پارس اتیلن
کیش میخرند
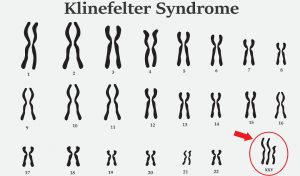
سندرم کلاین فلتر (47، XXY) یک تنوع کروموزومی در مردان است که در آن یک کروموزوم X اضافی وجود دارد که منجر به کاریوتایپ 47، XXY می شود. کروموزوم X اضافی معمولاً بر عملکرد فیزیکی، رشد عصبی، رفتاری و عصبی-شناختی تأثیر می گذارد. ویژگیهای فیزیکی رایج ممکن است شامل قد بلند، کاهش تون عضلانی، بیضههای کوچک (هیپوگنادیسم)، تاخیر در رشد بلوغ و فقدان ویژگیهای جنسی ثانویه مردانه مانند کاهش موهای صورت و بدن باشد. افزایش رشد سینه (ژنیکوماستی) ممکن است در اواخر بلوغ بدون مراقبت بیولوژیکی مناسب رخ دهد. با درمان مناسب، بروز ژنیکوماستی به طور معمول در کمتر از 10٪ از پسران رخ می دهد.
در پسران با (47، XXY) تغییرات زیادی در نمایه یا فنوتیپ رشد عصبی وجود دارد. ویژگیهای رایج شناختی و رفتاری ممکن است شامل تأخیر گفتار و زبان، ADHD و چالشهای عملکرد عاطفی و اجتماعی باشد. ویژگی های (47، XXY) معمولاً با کاهش سطح تستوسترون و افزایش سطح گنادوتروپین مرتبط است. درمان هورمونی اولیه (EHT) با سه تزریق ماهانه 25 میلی گرم. تستوسترون انانتات که معمولاً بین 4 تا 12 ماهگی تجویز می شود، ممکن است رشد مغز و نتایج رشد عصبی را بهینه کند. تستوسترون باید بر اساس ارزیابی با متخصص اطفال و متخصص غدد اطفال انجام شود.
48, XXYY Syndrome:
یک اختلال ژنتیکی است که در مردان رخ می دهد و با کروموزوم های جنسی اضافی در مقایسه با کاریوتایپ معمولی مردانه 46، XY مشخص می شود. 48، سندرم XXYY قبلاً بهعنوان گونهای از سندرم کلاین فلتر توصیف شده است، زیرا مردان مبتلا ویژگیهای فیزیکی مشابهی دارند (قد بلند و بیضههای کوچک و ناکارآمد)، اما ویژگیهای پزشکی و رشد عصبی پیچیدهتر از آنچه در سندرم XXY/Klinefelter مشاهده میشود، پیچیدهتر است.
Abetalipoproteinemia :
Abetalipoproteinemia یک اختلال ارثی نادر است که بر جذب چربی توسط روده و تحرک توسط کبد تأثیر می گذارد. ناتوانی در جذب چربی منجر به کمبود لیپیدها و انواع ویتامین های ضروری می شود. افراد مبتلا دچار ومشکلات عصبی، ضعف عضلانی، مشکل در راه رفتن، و ناهنجاری های خونی از جمله وضعیتی که در آن گلبول های قرمز بد شکل هستند (اکانتوسیتوز) و در نتیجه سطوح پایین گلبول های قرمز در گردش (کم خونی) را تجربه می کنند. افراد مبتلا همچنین ممکن است دچار دژنراسیون شبکیه چشم شوند که به طور بالقوه منجر به از دست دادن بینایی می شود، وضعیتی که به عنوان رتینیت پیگمنتوزا شناخته می شود. Abetalipoproteinemia به عنوان یک صفت اتوزومی مغلوب به ارث می رسد و در اثر جهش در ژن پروتئین انتقال تری گلیسیرید میکروزومی (MTTP) ایجاد می شود.
Ablepharon-Macrostomia Syndrome:
سندرم آبلفارون-ماکروستومی (AMS) یک اختلال ژنتیکی نادر است که با فقدان یا توسعه نیافته پلک ها (بلفارول یا میکرو بلفارون) و دهان باز (ماکروستومی) مشخص می شود. ویژگی ها عمدتاً صورت و پوست را درگیر می کند و به ندرت اندام های داخلی (احشاء) را درگیر می کند. علائم و نشانههای رایج علاوه بر یافتههای چشم و دهان شامل گوشهای کمتنظیم با لالههای گوش متصل، انحراف یا جوش خوردن انگشتها (سینداکتیلی یا کمپتوداکتیلی)، برآمدگی گونهها، نبودن یا بسیار کوچک نوک سینهها، پوست چروکیده و زائد، عدم وجود یا کمشدن موهای زائد است. و ناهنجاری های تناسلی سایر یافتههای کمتر گزارش شده شامل ناهنجاریهای ناف، تاخیر رشد و ناتوانی ذهنی است. AMS در دسته بیماریهایی به نام دیسپلازی اکتودرم (اختلالات ژنتیکی که شامل نقص در پوست، مو، ناخن، غدد عرق و/یا دندانها میشود) گروهبندی شده است، اما از آنجایی که بسیاری از ویژگیها شامل بافتهایی میشوند که از اکتودرم مشتق نشدهاند. بهتر است AMS را به عنوان یک سندرم ناهنجاری واقعی تعریف کنیم. AMS در اثر تغییرات (جهش) در ژن TWIST2 ایجاد می شود. الگوی وراثت اتوزومال غالب است و بیشتر موارد به صورت جهش خودبخودی ایجاد می شود، بنابراین به صورت پراکنده رخ می دهد.
جهش در TWIST2 همچنین باعث ایجاد سندرم باربر سی و سندرم ستلیز می شود که ویژگی های بسیار مشابهی دارند. در واقع، پیشنهاد شده است که این سه اختلال در واقع یک زنجیره را تشکیل می دهند.
Acanthocheilonemiasis:
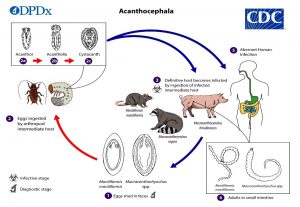
Acanthocheilonemiasis یک بیماری عفونی نادر استوایی است که توسط انگلی به نام Acanthocheilonema perstans ایجاد می شود که به گروهی از بیماری های انگلی معروف به بیماری های فیلاریال (نماتد) تعلق دارد. این انگل بیشتر در آفریقا یافت می شود. علائم عفونت ممکن است شامل قرمزی، خارش پوست (خارش)، درد شکم و قفسه سینه، درد عضلانی و نواحی تورم موضعی باشد. علاوه بر این، کبد و طحال ممکن است به طور غیر طبیعی بزرگ شوند (هپاتواسپلنومگالی). آزمایشات آزمایشگاهی همچنین ممکن است سطوح بالای غیرعادی گلبول های سفید خاص (ائوزینوفیلی) را نشان دهد. این انگل از طریق نیش مگس های کوچک (A. coliroides) منتقل می شود.
Aceruloplasminemia:
آسرولوپلاسمینمی یک اختلال ژنتیکی نادر است که با تجمع غیرطبیعی آهن در مغز و اندام های داخلی مختلف مشخص می شود. افراد مبتلا به علائم عصبی از جمله اختلالات شناختی و اختلالات حرکتی مبتلا می شوند. تخریب شبکیه و دیابت نیز ممکن است رخ دهد. علائم معمولاً در سنین بزرگسالی بین 20 تا 60 سالگی آشکار می شوند. آسرولوپلاسمینمی در اثر جهش در ژن سرولوپلاسمین (CP) ایجاد می شود. این جهش در الگوی اتوزومال مغلوب به ارث می رسد.
آسرولوپلاسمینمی به عنوان یک اختلال نورودژنراتیو با تجمع آهن در مغز (NBIA) طبقه بندی می شود. NBIA گروهی از اختلالات ارثی نادر است که با تجمع آهن در مغز مشخص می شود. آسرولوپلاسمینمی نیز به عنوان یک اختلال اضافه بار آهن طبقه بندی می شود.
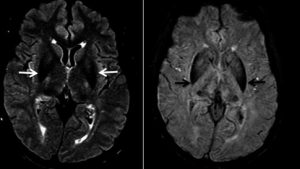
تصویر سمت چپ : تصاویر رزونانس مغناطیسی مغز با سیگنال هایپو شدید برجسته در ناحیه عقده های پایه های سفید
تصویر سمت راست : تصاویر وزنی حساسیت رزونانس مغناطیسی مغز (SWI) که رسوب آهن را در گانگلیون های پایه (فلش های سیاه) نشان می دهد.
Achondrogenesis:
آکندروژنز گروهی از دیسپلازی های اسکلتی نادر است که با کوتاه شدن شدید دست ها و پاها در رابطه با تنه، رشد غیر طبیعی دنده ها، مهره ها و سایر ناهنجاری های اسکلتی مشخص می شود. مشکلات سلامتی مرتبط با این بیماری ها تهدید کننده زندگی هستند و اکثر نوزادان مبتلا مرده به دنیا می آیند یا اندکی پس از تولد به دلیل نارسایی تنفسی می میرند. همه انواع آکندروژنز شرایط ژنتیکی هستند. نوع IA و نوع IB، اختلالات اتوزومال مغلوب هستند، در حالی که آکندروژنز نوع II یک اختلال اتوزومال غالب است. همه انواع آکندروژنز، دیسپلازی های اسکلتی بسیار شدید هستند که معمولاً با معاینه اولتراسوند قبل از تولد در اوایل هفته 14-17 سن حاملگی تشخیص داده می شوند.
Acidemia, Methylmalonic:
اسیدمی های متیل مالونیک اسیدمی های آلی هستند که در اثر نقص آنزیمی در متابولیسم چهار آمینو اسید (متیونین، ترئونین، ایزولوسین و والین) ایجاد می شوند. این منجر به افزایش غیرطبیعی اسید در خون (آکادمی) و بافت های بدن می شود. در شکل حاد، خواب آلودگی، کما و تشنج ممکن است رخ دهد. عقب ماندگی ذهنی یک پیامد طولانی مدت است. این اختلال ممکن است ناشی از کمبود یک یا چند آنزیم متیل مالونیل کوآ موتاز، متیل مالونیل راسماز یا آنزیم های مصنوعی آدنوزیل کوبالامین باشد. دفع متیل مالونات، محصول متابولیسم اسیدهای آمینه، در ادرار به طور غیر طبیعی زیاد است و بنابراین نشانگر این اختلال است. تمام اسیدمی های آلی شناخته شده به عنوان صفات اتوزومال مغلوب به ارث می رسند.
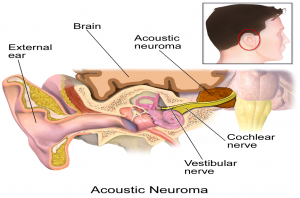
Acoustic Neuroma:
نوروم آکوستیک که به عنوان شوانوم دهلیزی نیز شناخته می شود، یک رشد خوش خیم (غیر سرطانی) نادر است ،که در عصب هشتم جمجمه ایجاد می شود. این عصب از گوش داخلی به مغز می رود و مسئول شنوایی و تعادل (تعادل) است. اگرچه هیچ الگوی استاندارد یا معمولی برای ایجاد علائم وجود ندارد، کاهش شنوایی در یک گوش (یک طرفه) علامت اولیه تقریباً 90 درصد از افراد مبتلا است. یافته های رایج دیگر شامل صدای زنگ در گوش (وزوز گوش) و سرگیجه یا عدم تعادل است. علائم نوروم آکوستیک از فشار تومور به عصب هشتم جمجمه ای و اختلال در توانایی آن در انتقال سیگنال های عصبی به مغز رخ می دهد. نوروم آکوستیک سرطانی نیست (بدخیم). به سایر قسمت های بدن سرایت نمی کند. دلیل ایجاد نوروم آکوستیک ناشناخته است.
Acquired Aplastic Anemia:
کم خونی آپلاستیک اکتسابی یک اختلال خونی نادر و جدی است که به دلیل نارسایی مغز استخوان در تولید سلول های خونی است. مغز استخوان ماده اسفنجی است که در مرکز استخوانهای بدن، در بزرگسالان عمدتاً ستون فقرات، لگن و استخوانهای بزرگ پاها یافت میشود. مغز استخوان حاوی سلول های بنیادی خونساز است. سلولهای بنیادی میتوانند سلولهای بنیادی بیشتری تولید کنند (خود نوسازی) و همچنین تمایز و تکثیر میشوند و گلبولهای قرمز (گلبولهای قرمز)، گلبولهای سفید (لوکوسیتها) و پلاکتها را تشکیل میدهند. در کم خونی آپلاستیک اکتسابی، فقدان تقریباً کامل سلول های بنیادی خونساز منجر به کاهش سطح گلبول های قرمز و سفید و پلاکت ها می شود (پانسیتوپنی). علائم کم خونی آپلاستیک علائم کم خونی، خونریزی و عفونت است. اگرچه نارسایی مغز استخوان می تواند ثانویه به سایر اختلالات رخ دهد، اکثر کم خونی آپلاستیک به این دلیل است که سیستم ایمنی به اشتباه مغز استخوان را هدف قرار می دهد (خود ایمنی). در واقع، اکثر بیماران می توانند به درمانی که سیستم ایمنی را سرکوب می کند، معمولاً ATG و سیکلوسپورین، پاسخ دهند.
Acquired Lipodystrophy:
لیپودیستروفی اکتسابی یک اصطلاح کلی برای انواع لیپودیستروفی است که ارثی نیستند، بلکه در برخی از مراحل زندگی به دست می آیند. لیپودیستروفی های اکتسابی علت ژنتیکی مستقیمی ندارند، بلکه ممکن است عوامل مختلفی در آن دخیل باشند. لیپودیستروفی اکتسابی می تواند در اثر داروها، خودایمنی یا به دلایل ناشناخته (ایدیوپاتیک) ایجاد شود. انواع فرعی لیپودیستروفی اکتسابی شامل لیپودیستروفی عمومی اکتسابی (سندرم لارنس)، لیپودیستروفی نسبی اکتسابی (سندرم باراکور-سیمونز)، لیپودیستروفی موضعی و لیپودیستروفی ناشی از آنتی رتروویروسی فعال بالا است که ممکن است در افراد آلوده به HIV ایجاد شود که تحت یک شکل خاص درمان قرار می گیرند. شروع اشکال اکتسابی لیپودیستروفی می تواند در دوران کودکی، نوجوانی یا بزرگسالی رخ دهد. افراد مبتلا دچار از دست دادن مشخصه چربی بدن (بافت چربی) می شوند که بر نواحی خاصی از بدن، به ویژه بازوها، پاها، صورت، گردن، و قفسه سینه یا نواحی قفسه سینه تأثیر می گذارد. در برخی موارد، عوارض متابولیک مرتبط با مقاومت به انسولین ممکن است ایجاد شود. چنین عوارضی شامل ناتوانی در تجزیه گلوکز (عدم تحمل گلوکز)، افزایش سطح تری گلیسیرید (نوعی چربی) در خون (هیپرتری گلیسیریدمی) و دیابت است. علائم اضافی مانند تجمع چربی در کبد (کبد چرب یا استئاتوز کبدی) نیز ممکن است رخ دهد.
Acrocallosal Syndrome, Schinzel Type:
سندرم آکروکالوزال، نوع شینزل یک اختلال ژنتیکی نادر است که در بدو تولد آشکار می شود (مادرزادی). علائم و یافته های مرتبط ممکن است متغیر باشد، از جمله در میان اعضای آسیب دیده یک خانواده (خویشاوندان). با این حال، این اختلال معمولاً با توسعه نیافتگی (هیپوپلازی) یا فقدان (آژنزی) نوار ضخیم رشته های عصبی که به دو نیمکره مغز می پیوندند (corpus callosum) و عقب ماندگی ذهنی متوسط تا شدید مشخص می شود. علاوه بر این، بسیاری از افراد مبتلا ناهنجاری های جمجمه و ناحیه صورت (جمجمه صورت) و/یا ناهنجاری های متمایز انگشتان دست و پا (اعداد) دارند. ناهنجاریهای مشخصه جمجمهصورتی ممکن است شامل سر غیرمعمول بزرگ (ماکروسفالی) با پیشانی برجسته، چشمهای با فاصله زیاد (هیپرتلوریسم چشمی)، چینهای پلک به سمت پایین (شکافهای کف دست)، بینی کوچک با پل بینی پهن باشد. و گوش های بد شکل (دیسپلاستیک). بیشتر افراد مبتلا همچنین دارای ناهنجاریهای دیجیتالی مشخصی هستند، مانند وجود انگشتان دست و پا (پلی داکتیلی) اضافی و تار شدن یا فیوژن (سینداکتیلی) برخی از انگشتان. همچنین ممکن است ناهنجاریهای فیزیکی اضافی نیز وجود داشته باشد، از جمله تاخیر در رشد، که منجر به کوتاهی قد میشود. اگرچه توارث اتوزومال مغلوب پیشنهاد شده است، سندرم آکروکالوزال اغلب به دلایل ناشناخته (به صورت پراکنده) به طور تصادفی رخ می دهد.
Wyburn-Mason Syndrome:
سندرم وایبرن میسون یک اختلال نادر غیر ارثی است که در بدو تولد وجود دارد (مادرزادی). نوزادان مبتلا ناهنجاری های شریانی وریدی (AVMs) دارند که ناهنجاری های رشدی هستند که بر رگ های خونی، به ویژه شریان ها، سیاهرگ ها و مویرگ ها تأثیر می گذارد. سرخرگ ها معمولا خون غنی از اکسیژن را از قلب به سلولهای بدن میبرند، در حالی که سیاهرگها خون کمبود اکسیژن را برای تبادل اکسیژن و دی اکسید کربن به قلب و ریهها میرسانند. شبکه رگهای خونی بسیار ریز (مویرگها) که به طور معمول شریانها و سیاهرگها را به هم متصل میکنند ممکن است وجود نداشته باشند و سرخرگها و سیاهرگها ممکن است مستقیماً به یکدیگر متصل شده و یک ناهنجاری را ایجاد کنند. بدون مویرگ ها، ممکن است به دیواره سرخرگ ها و وریدها آسیب وارد شود و باعث جریان خون غیرطبیعی و زیاد و نشت و عدم جریان خون بیشتر در پایین دست شود. AVMهای بزرگتر ممکن است از توده درهم پیچیده عروق خونی غیرطبیعی یا بدشکل (نیدوس) تشکیل شده باشند. AVM های مرتبط با سندرم وایبرن میسون معمولا در چشم ها (شبکیه) و مغز یافت می شوند. علت دقیق سندرم Wyburn-Mason ناشناخته است، اگرچه این فرضیه وجود دارد که در طول رشد اولیه، سلولهای پیش ساز رگهای خونی حرکت غیرطبیعی (مهاجرت) دارند که باعث توسعه غیرطبیعی بعدا میشود.
Zellweger Spectrum Disorders:
اختلالات طیف زلوگر (ZSD) گروهی از اختلالات نادر، ژنتیکی و چند سیستمی هستند که زمانی تصور میشد موجودیتهای جداگانهای باشند. این اختلالات در حال حاضر به عنوان عبارات مختلف (انواع) یک فرآیند بیماری به دلیل پایه بیوشیمیایی مشترک آنها طبقه بندی می شوند. در مجموع، آنها طیف یا زنجیره ای از بیماری را تشکیل می دهند. شدیدترین شکل این اختلالات قبلاً به عنوان سندرم زلوگر، شکل میانی به عنوان آدرنولوکودیستروفی نوزادان و اشکال خفیف تر به عنوان بیماری رفسوم نوزادی یا سندرم هایملر، بسته به تظاهرات بالینی، نامیده می شد. ZSD می تواند بیشتر اندام های بدن را تحت تاثیر قرار دهد. نقایص عصبی، از دست دادن توان عضلانی (هیپوتونی)، کاهش شنوایی، مشکلات بینایی، اختلال عملکرد کبد و ناهنجاری های کلیوی یافته های رایج هستند. ZSD اغلب در اوایل دوران نوزادی منجر به عوارض شدید و تهدید کننده زندگی می شود. برخی از افراد با اشکال خفیف تر تا بزرگسالی زندگی کرده اند. ZSD در الگوی اتوزومال مغلوب به ارث می رسد.
Wandering Spleen:
طحال سرگردان مادرزادی یک نقص بسیار نادر است که به طور تصادفی توزیع شده است که با فقدان یا ضعف یک یا چند رباط که طحال را در موقعیت طبیعی خود در سمت چپ بالای شکم نگه می دارند مشخص می شود. این اختلال منشا ژنتیکی ندارد. به جای رباط ها، طحال توسط یک بافت ساقه مانند که با رگ های خونی تامین می شود (پدیکول عروقی) متصل می شود. اگر ساقه در طول حرکت طحال پیچ خورده باشد، جریان خون ممکن است قطع یا مسدود شود (ایسکمی) تا حد آسیب شدید به عروق خونی (انفارکتوس). از آنجایی که نمی توان آن را در جای خود نگه داشت یا چیزی وجود ندارد، طحال در قسمت تحتانی شکم یا لگن “سرگردان” می شود، جایی که ممکن است با یک توده شکمی ناشناس اشتباه گرفته شود.
طحال عضو کوچکی است که در سمت چپ بالای شکم قرار دارد. طحال مواد غیر ضروری یا خارجی را از بین می برد یا فیلتر می کند، سلول های خونی فرسوده را می شکند و از بین می برد و گلبول های سفید تولید می کند که به بدن در مبارزه با عفونت کمک می کند. علائم طحال سرگردان معمولاً علائمی است که با اندازه غیرطبیعی طحال (سپلنومگالی) یا موقعیت غیرعادی طحال در شکم همراه است. بزرگ شدن اغلب نتیجه پیچ خوردگی (پیچ خوردگی) شریان ها و وریدهای طحال یا در برخی موارد تشکیل لخته خون (انفارکتوس) در طحال است.
طحال سرگردان “اکتسابی” ممکن است در دوران بزرگسالی به دلیل صدمات یا سایر شرایط زمینهای که ممکن است رباطهایی را که طحال را در موقعیت طبیعی خود نگه میدارند ضعیف کند (به عنوان مثال، بیماری بافت همبند یا بارداری) رخ دهد.
Trigeminal Neuralgia:
نورالژی سه قلو (TN)، همچنین به عنوان tic douloureux شناخته می شود، یک اختلال در عصب جمجمه پنجم (عصب سه قلو) است. این اختلال گاهی به نوع 1 و نوع 2 تقسیم می شود. TN نوع 1 (TN1) با حملات درد شدید و کوبنده که دهان، گونه، بینی و/یا سایر نواحی یک طرف صورت را تحت تاثیر قرار می دهد مشخص می شود. TN نوع 2 (TN2) با درد با شدت کمتر، اما درد مبهم یا سوزش ثابت مشخص می شود. هر دو نوع درد می تواند در یک فرد حتی در یک زمان ایجاد شود. در برخی موارد، درد می تواند طاقت فرسا و ناتوان کننده باشد. در صورت عدم درمان، TN می تواند تأثیر عمیقی بر کیفیت زندگی فرد داشته باشد. در بیشتر موارد، TN1 به دلیل فشار دادن رگ خونی به عصب سه قلو ایجاد می شود، اما گاهی اوقات هیچ علت زمینه ای نمی توان شناسایی کرد (ایدیوپاتیک). TN2 می تواند به دلیل فشرده سازی عصب سه قلو، ایدیوپاتیک باشد یا به دلیل یک علت زمینه ای شناخته شده مانند تومور یا مولتیپل اسکلروزیس رخ دهد. مشخص نیست که چرا یک نفر علائم TN1 در مقابل TN2 را نشان می دهد. ممکن است به دلیل تعداد عروق (به عنوان مثال شریان ها، وریدها) یا میزان فشرده سازی باشد. TN معمولاً از طریق داروها، جراحی یا تزریق قابل کنترل است.
Paraneoplastic Neurologic Syndromes :
سندرمهای عصبی پارانئوپلاستیک (PNS) گروهی از شرایطی هستند که بر سیستم عصبی (مغز، نخاع، اعصاب و/یا ماهیچهها) در بیماران مبتلا به سرطان تأثیر میگذارند. اصطلاح “پارانئوپلاستیک” به این معنی است که سندرم عصبی توسط خود تومور ایجاد نمی شود، بلکه توسط واکنش های ایمونولوژیکی که تومور ایجاد می کند ،ایجاد می شود. اعتقاد بر این است که سیستم ایمنی طبیعی بدن تومور را به عنوان یک حمله تفسیر می کند. هنگامی که این اتفاق می افتد، سیستم ایمنی یک پاسخ ایمنی ایجاد می کند و از آنتی بادی ها و لنفوسیت ها برای مبارزه با تومور استفاده می کند. نتیجه نهایی این است که سیستم ایمنی خود بیمار می تواند باعث آسیب جانبی به سیستم عصبی شود که گاهی اوقات می تواند شدید باشد. در بسیاری از بیماران، پاسخ ایمنی می تواند باعث آسیب به سیستم عصبی شود که بسیار بیشتر از آسیب وارد شده به تومور است. اثرات PNS می تواند به طور کامل از بین برود، اگرچه می تواند اثرات دائمی نیز داشته باشد.
Sweet Syndrome:
سندرم شیرین یک اختلال نادر است که با تب و شروع ناگهانی به صورت پوستی مشخص می شود که شامل برآمدگی ها یا ضایعات حساس، قرمز یا قرمز مایل به آبی است. این ضایعات معمولا در بازوها، پاها، تنه، صورت یا گردن ایجاد می شوند. در برخی موارد، سیستمهای اضافی بدن از جمله سیستم اسکلتی-عضلانی مانند التهاب مفاصل (آرتریت)، چشمها مانند التهاب ملتحمه یا غشایی که چشمها را میپوشاند (کانژنکتیویت) و اندامهای داخلی ممکن است درگیر شوند. در اکثر افراد مبتلا، این اختلال به خودی خود و بدون هیچ دلیل شناخته شده ای رخ می دهد (سندرم شیرین ایدیوپاتیک). این همچنین به عنوان سندرم شیرین کلاسیک شناخته می شود. در موارد کمتر، این اختلال میتواند با یک سرطان زمینهای (بدخیم)، معمولاً سرطان خون (هماتولوژیک) مانند انواع خاصی از لوسمی مرتبط باشد. این به عنوان سندرم شیرین مرتبط با بدخیمی شناخته می شود. این اختلال همچنین می تواند به عنوان یک واکنش به مصرف برخی داروها، به ویژه دارویی به نام فاکتور تحریک کننده کلونی گرانولوسیتی منجر شود. این به عنوان سندرم شیرین ناشی از دارو شناخته می شود. سندرم شیرین با کورتیکواستروئیدها درمان می شود.
Ovarian Cancer:
سرطان تخمدان به سرطانی اطلاق میشود که در اندامهای تناسلی بادام شکل در زنان ایجاد میشود که در آن تخمکها یا تخمدانها (تخمدان)، در هر یک از دو لولهای که از طریق آن تخمکها از تخمدان به رحم میروند (لولههای فالوپ) ایجاد میشود. یا در غشایی که لگن و حفره شکمی را می پوشاند و تمام اندام های شکمی را می پوشاند (صفاق). معمولاً در مراحل اولیه هیچ علامتی وجود ندارد (بدون علامت). در نتیجه سرطان تخمدان معمولا تا مرحله پیشرفته تشخیص داده نمی شود. علائم رایجی که می تواند ایجاد شود شامل تورم یا نفخ شکم، کاهش وزن ناخواسته، یا تغییر در عادات روده از جمله یبوست است. علت سرطان تخمدان چند عاملی است، به این معنی که عوامل متعددی که با هم رخ می دهند برای ایجاد سرطان ضروری هستند. این عوامل شامل عوامل ژنتیکی، ایمونولوژیک و محیطی است. حدود 20 درصد از تمام زنان مبتلا به سرطان تخمدان دارای تنوع در یکی از دو ژن به نام BRCA1 یا BRCA2 هستند. سرطان تخمدان مرتبط با BRCA به طور خاص در این گزارش مورد بحث قرار نگرفته است. NORD گزارش جداگانه ای در مورد سرطان تخمدان ناشی از تغییرات در این دو ژن دارد که با عنوان سندرم ارثی پستان و سرطان تخمدان (HBOC) نامیده می شود.
سرطان تخمدان یک اصطلاح غیر اختصاصی برای گروهی از تومورها است. با این حال، بیش از 90٪ سرطان تخمدان از سلول های اپیتلیال منشأ می گیرد. این سلول ها بیشتر سطوح داخلی و خارجی بدن و اندام های آن را می پوشانند. سرطان اپیتلیال تخمدان ها، لوله های فالوپ و صفاق رفتار و ویژگی های مشابهی دارند. این سرطان های نزدیک به هم اغلب با هم گروه بندی می شوند و می توانند به عنوان سرطان تخمدان اپیتلیال یا EOC نامیده شوند. این گزارش در درجه اول به سرطان اپیتلیال تخمدان (کارسینوم) می پردازد. بیشتر موارد EOC سرطان سروز درجه بالا هستند. اکنون اعتقاد بر این است که اکثر کارسینوم های سروزی درجه بالا از نوک لوله های فالوپ منشا می گیرند. زیرگروه های اضافی EOC عبارتند از: کارسینوم سروزی با درجه پایین، موسینوس، سلول شفاف، اندومتروئید و تمایز نیافته که در آن سلول های سرطانی بسیار نابالغ هستند و شبیه سلول های بافتی که سرطان در آن شروع می شود، نیستند.
انواع دیگر سرطان تخمدان عبارتند از تومورهای استرومایی، که در آن سرطان در بافت تخمدانی که حاوی سلولهای تولیدکننده هورمون است، و تومورهای سلول زایا، که در آن سرطان در سلولهای تولیدکننده تخمک ایجاد میشود، ایجاد میشود. به تومورهای استرومایی تومورهای بند ناف جنسی – استرومایی نیز گفته می شود و حدود 7 درصد از زنان مبتلا به سرطان تخمدان را تشکیل می دهند. انواع تومور استرومایی شامل گرانولوزا، گرانولوزا-تکا و سلول سرتولی لیدیگ است. تومورهای سلول زایای بسیار نادر هستند و در زنان جوانتر (حدود 80 درصد در زنان زیر 30 سال) رخ میدهند. انواع تومورهای سلول زاینده عبارتند از تراتوم، دیس ژرمینوما، تومور سینوس اندودرم و کوریوکارسینوما.
 Call Us : (+98 21) 88 20 20 60
Call Us : (+98 21) 88 20 20 60